Alexis St-Gelais – Vulgarisation
[Des ajouts au texte initial ont été faits le 23 mars 2019, et sont notés en vert]
Une partie importante de l’exercice de la chimie analytique consiste à sélectionner le détecteur qui convient à l’objectif de l’analyse. Chaque détecteur présente en effet ses pour et ses contre. Dans le secteur des huiles essentielles, FID et MS (en mode de mesure des ions totaux) sont tous les deux fréquemment utilisés, et pas toujours à bon escient. Malheureusement, il est rare qu’on explique aux clients et aux consommateurs la différence entre les deux, et on les présente même faussement comme étant équivalents. Je tâcherai d’y remédier ici. Le tableau 1 compare quelques caractéristiques des deux détecteurs.
Tableau 1. Caractéristiques respectives des détecteurs
FID et MS en chromatographie en phase gazeuse
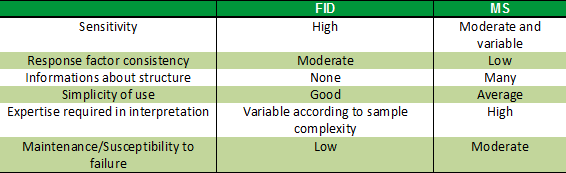
De manière générale, le MS doit être préféré pour l’analyse qualitative (identification des molécules en cas de doute), et le FID pour l’analyse quantitative (établir le pourcentage de concentration de chaque composé dans l’huile essentielle). On voit cependant souvent des rapports d’analyse d’huiles essentielles présentant des pourcentages de concentration à partir d’une seule détection sur MS, ce qui n’est pas une bonne pratique (2).
D’abord, il faut savoir que les chromatogrammes obtenus par MS sont généralement des « total ion chromatograms » (TIC), donc la somme du nombre de fragments mesurés pour une molécule. Si une molécule se fragmente facilement, son signal en TIC sera intense, alors qu’une molécule « robuste » produisant peu d’ions fournira un signal faible. Cela entraîne des disparités entre les facteurs de réponse* observés (1,2). Comme je l’ai mentionné dans un billet précédent, le facteur de réponse en FID n’est pas non plus égal pour toutes les molécules, mais il est plus similaire, ce qui réduit les biais de quantification. Observons concrètement ce qu’il en est.
J’ai préparé une solution d’une série de molécules volatiles de différents types à des concentrations connues. J’ai donc pu déterminer leur teneur en pourcentage de la masse totale de composés volatils de manière absolue (tableau 2). J’ai ensuite injecté le mélange avec la même programmation de température sur GC-MS et GC-FID et intégré les chromatogrammes obtenus. J’ai ensuite calculé l’écart entre le pourcentage lu lors de l’analyse et le pourcentage réel.
Tableau 2. Comparaison de la performance de quantification des
détecteurs FID et MS pour un mélange connu de composés volatils
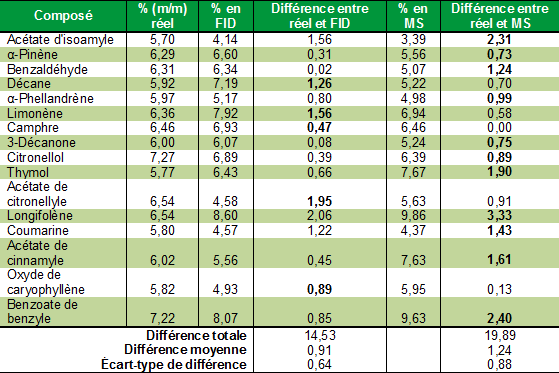
En gras: Erreur de quantification plus importante que sur l’autre détecteur
On remarque que le MS produit dans l’ensemble plus de distorsion de la composition réelle du mélange que le FID. La différence totale est supérieure, tout comme la différence moyenne pour un composé. Enfin, l’écart-type plus élevé indique que cette variation est plus étalée (donc plus imprévisible) en MS. D’autres recherches abondent dans le même sens (1,2). Le MS peut aussi être fiable, mais cela requiert des précautions plus lourdes (3) et une bonne calibration, ce qui s’applique mal aux huiles essentielles de composition très variable. Cicchetti, Merle et Chaintreau, à la suite d’une expérience similaire, ont conclu en 2008 que la quantification par pourcentage en MS « a peu de chances d’être reproductible, et qu’elle est donc inappropriée pour décrire la composition d’une échantillon » (2).
On constate qu’aucune des deux méthodes n’est entièrement fidèle à la composition du mélange. Cela peut s’expliquer en partie par les nombreux types de molécules que j’y ai mises : alcanes, terpènes, composés phénoliques, esters s’y côtoient. C’est une diversité que l’on trouve rarement dans les huiles essentielles.
J’insiste de nouveau sur le fait que le FID est comparativement plus fiable que le MS, mais n’est pas parfait (aucun détecteur ne l’est). On l’utilise par convention, et pour faire des comparaisons fiables entre les analyses. La détection par FID est celle préconisée par les normes AFNOR, qui servent de référence dans le secteur de l’analyse des composés volatils (4). Nous verrons dans de prochains billets comment nous pouvons améliorer davantage les résultats obtenus en GC-FID à l’aide de facteurs de correction, ou mieux encore avec une calibration en bonne et due forme.
*Le facteur de réponse désigne la constante par laquelle il faut diviser le signal lu par un détecteur pour obtenir la quantité d’une molécule en masse. Par exemple, si 2,0 µg d’une molécule passant par un détecteur donnent un signal de 50000 unités (absorbance de lumière, courant électrique, etc.), le facteur de réponse est de 25 000 unités/µg.
Références
(1) Bicchi, C., Liberto, E., Matteodo, M., Sgorbini, B., Mondello, L., d’Acampora Zellner, B., Costa, R., Rubiolo, P. Quantitative analysis of essential oils: a complex task. Flavour Fragr. J. 2008, 23, 382–391.
(2) Cicchetti, E., Merle, P., Chaintreau, A. Quantitation in gas chromatography: usual practices and performances of a response factor database. Flavour Fragr. J. 23, 450-459.
(3) Dodds, E., McCoy, M., Rea, L., Kennish, J. M. Gas Chromatographic Quantification of Fatty Acid Methyl Esters: Flame Ionization Detection vs. Electron Impact Mass Spectrometry. Lipids 2005, 40(4), 419-428.
(4) AFNOR. Directives générales concernant les profils chromatographiques – Partie 2: Utilisation des profils chromatographiques des échantillons d’huiles essentielles, ISO 11024-2:1999(F).
